An der Augenklinik des LMU Klinikums München wurde erstmals ein 13 Monate alter Junge erfolgreich mit einer Gentherapie gegen Leber’sche Kongenitale Amaurose (LCA) behandelt – einer der jüngsten Patienten weltweit. Der Junge leidet an der speziellen LCA2-Form der Erkrankung. Bei der Therapie wird ein defektes RPE65-Gen durch eine funktionstüchtige Genkopie ersetzt, die direkt unter die Netzhaut injiziert wird. Das von diesem Gen kodierte Enzym sorgt dafür, dass ein für das Sehen notwendiger Stoff – ein lichtempfindliches Vitamin-A-Derivat – immer wieder erneuert wird. Der Eingriff verlief komplikationslos; bereits drei Wochen später zeigte der Patient eine deutliche Sehverbesserung.
LCA steht für Leber’sche Kongenitale Amaurose. Sie ist keine einzelne Erkrankung, sondern eine Gruppe seltener, angeborener Netzhauterkrankungen, die alle zu einem ähnlichen Krankheitsbild führen und lange Zeit als unheilbar galten. Häufig sind genetische Veränderungen die Ursache. Eine spezielle Form, die sogenannte LCA2, entsteht durch Mutationen im sogenannten RPE65-Gen. Dieses Gen ist in den retinalen Pigmentepithelzellen der Netzhaut aktiv und spielt eine wichtige Rolle im Sehprozess. Es sorgt dafür, dass ein für das Sehen notwendiger Stoff – ein lichtempfindliches Vitamin-A-Derivat – immer wieder erneuert wird. Ist diese Funktion gestört, kann das Auge Licht nicht mehr richtig verarbeiten. Betroffene Patientinnen und Patienten leiden daher schon früh unter stark eingeschränktem Sehvermögen und ausgeprägter Nachtblindheit. Im weiteren Verlauf kann sich die Sehfähigkeit weiter verschlechtern – bis hin zur Erblindung.
Eingeschleuste intakte RPE65-Genkopien für eine bessere Sehkraft
Ist das RPE65-Gen defekt, kann seit einigen Jahren in der Netzhaut ein gesundes Gen hinzugefügt (supplementiert) werden. Hierfür wird das funktionstüchtige Gen im Rahmen eines einmalig mikrochirurgischen Eingriffs nach Entfernung des Glaskörpers unter die Netzhaut am Augenhintergrund injiziert und dann in einer Art Virus-Taxi in die krankhaften Zellen der Netzhaut eingeschleust. Der therapeutische Effekt: Die defekten Zielzellen nehmen ihre Funktion wieder auf und sorgen so dafür, dass der physiologische Sehzyklus wiederhergestellt wird. „Auf diese Weise können wir nicht nur den Krankheitsverlauf bremsen, sondern tatsächlich verloren gegangene Funktionen zurückgewinnen – und das möglicherweise dauerhaft,“ sagt Prof. Dr. Siegfried Priglinger. Der Direktor der Augenklinik am LMU Klinikum München führte die damals neu zugelassene Gentherapie 2019 als Erster in Deutschland durch.
Je früher behandelt wird, desto besser
Die klinische Symptomatik einer LCA beginnt oft schon nach der Geburt. Dennoch bleibt die Erkrankung in vielen Fällen lange unerkannt. So ist es auch Magnus und seinen Eltern ergangen, die erst nach einer Ärzteodyssee die Diagnose LCA erhielten. 2022 wurde er von Professor Priglinger in der Augenklinik operiert. Der Behandlungserfolg hält bis heute an und hat die Lebensqualität des inzwischen Zehnjährigen deutlich verbessert.
Nun operierte Professor Priglinger erstmals einen 13-monatigen Jungen mit LCA. Er ist einer von zwei jüngsten kleinen Patienten weltweit, bei dem die Gentherapie angewendet wurde. Der Eingriff an einem so kleinen Auge bedeutet selbst für den erfahrenen Operateur eine besondere Herausforderung. Für die LCA gilt jedoch: Die Prognose ist am besten, wenn die Gentherapie so früh wie möglich erfolgt. Denn dann ist die Chance besonders groß, dass noch genug vitales Netzhautgewebe vorhanden ist. Ist die Degeneration jedoch zu weit fortgeschritten, greift die Gentherapie nicht mehr.
Der kleine Wares hat den Eingriff am ersten Auge bestens überstanden – ohne nennenswerte Beeinträchtigungen. „Das ist ein weiteres Plus der Behandlung: dass sie im Allgemeinen sehr gut verträglich und nebenwirkungsarm ist“, sagt Professor Priglinger. Bereits nach drei Wochen beobachteten die Eltern eine deutliche Verbesserung des Sehvermögens bei ihrem Sohn: „Plötzlich konnte er erstmals zielgerichtet nach dem Handy greifen“, freut sich der Vater. Letzte Woche erfolgte die OP am zweiten Auge des kleinen Patienten, die Professor Priglinger im Rahmen des Kongresses VM Retina Meeting München als Live-OP durchführte. Auch dieser Eingriff verlief vollkommen problemlos. Wares kann nun auf eine deutlich verbesserte Sehfähigkeit hoffen – und damit auf ein weitgehend beschwerdefreies Leben.
Positive Bilanz
Inzwischen können Professor Priglinger und sein Team auf sieben Jahre Erfahrung mit der Gentherapie zurückblicken – und die Bilanz ist rundum positiv: „Zwar lässt sich das Sehvermögen meist nicht vollständig wiederherstellen, doch für Kinder, die in Dunkelheit und bei schlechten Lichtverhältnissen kaum noch etwas sehen konnten, bedeutet diese Behandlung einen Quantensprung.“
Weitere Gentherapien für andere Netzhauterkrankungen finden sich bereits in klinischer Erprobung. „Dies unterstreicht die Bedeutung der Präzisionsmedizin bei der Behandlung seltener vererbbarer Augenleiden. Denn Gentherapien eröffnen die Aussicht, bislang unheilbare Augenerkrankungen zu therapieren und so zu verhindern, dass Menschen erblinden“, so Professor Priglinger.
Wissenschaftliche Ansprechpartner:
Professor Dr. Siegfried Priglinger
Direktor der Augenklinik des LMU Klinikums München
Mathildenstraße 8, 80336 München
Tel. +49 (0) 89 4400-53811
E-Mail: augenklinik.direktion@med.uni-muenchen.de
Dr. Maximilian-Joachim Gerhardt
Facharzt in oberärztlicher Funktion
Augenklinik des LMU Klinikums München
Tel: +49 (0) 89 4400-53770
E-Mail: maximilian.gerhardt@med.uni-muenchen.de
Quelle: LMU
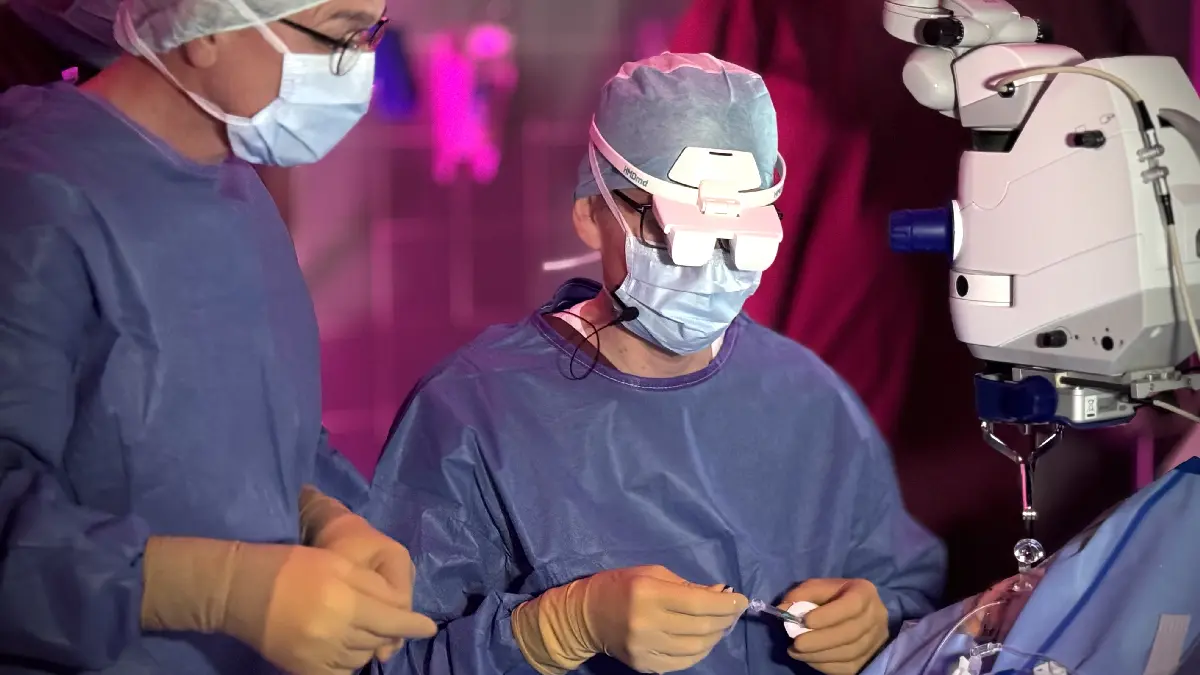
Bild: Prof. Dr. Siegfried Priglinger (rechts im Bild) während der Operation am LMU Klinikum, die im Rahmen des Kongresses VM Retina Meeting München live übertragen wurde. Foto: LMU Klinikum / Siegfried Priglinger